
The latest book in Robert Jordan's Wheel of Time series, The Gathering Storm has appeared in B&N, although my last understanding is that it wasn't due 'till 11/03. I don't have time for a real review, and by the time I've created one everybody interested will probably have read the book, but herewith a few notes.
For those unfamiliar with the series, I have linked to the Wiki page, but haven't read it. It may not be completely reliable, as the series may be a contentious subject and Wiki sometimes has problems with these. However, it should give you a general idea. If you want to become familiar, I suggest starting with the first book in the series ("Eye of the World") and reading forwards. Don't rely on any sort of summaries. Watch out also for some half-sized books (for children) which split the first few volumes into smaller chunks. A careful perusal of the Tor website or Wiki should allow you to find which is which.
Jordan's posthumous co-writer, Brandon Sanderson, says in the preface, introduction, or whatever (I don't have the book with me as I write) that we should consider this the first 1/3 of the final book of the series ("A Memory of Light"), and I strongly agree.
He also mentions that he has written in his own style, rather than trying to imitate Jordan. I find the writing itself rather similar, however he (IMO) takes a different approach to simultaneity, with different scenes much more separated in time than Jordan. (I may be mistaken about this, I just don't have time to go back and double check. And in any case, this doesn't include the first and last chapters of previous books, whose scenes have always been somewhat out of sync.)
I found this book much more satisfying than the last few, in the sense that many more issues are being resolved than opened. This was to be expected, but I can confirm it.
I'm not going to discuss plot details, or even list which issues have been resolved, but I will say that it's my impression that there are far fewer surprises here than in previous books. That doesn't mean that any issues were resolved in precisely the ways I had anticipated, but that in general the resolution fit within my broader expectations.
I strongly recommend reading it if you're following the series, and I can say that IMO Sanderson is doing a good enough job that we can expect to enjoy the finalization of the series almost as much as if Jordan had done it himself.
Read more!
Sunday, November 1, 2009
Friday, October 2, 2009
Ardipithecus ramidus Illuminates Human Origins (Science Mag: Open Access)
I'm really short on time, but readers need to know about the October 2 edition of Science Magazine, which:
presents 11 papers, authored by a diverse international team, describing an early hominid species, Ardipithecus ramidus, and its environment. These 4.4 million year old hominid fossils sit within a critical early part of human evolution, and cast new and sometimes surprising light on the evolution of human limbs and locomotion, the habitats occupied by early hominids, and the nature of our last common ancestor with chimps.I haven't had time to read much of it yet, but what I've seen is very suggestive. Read more!
Science is making access to this extraordinary set of materials FREE (non-subscribers require a simple registration).
Monday, September 28, 2009
Encephalon #76 Is Up
Here.
Looks like a great line-up.
BTW, I'm really busy with outside projects right now, so big posts are going to be sort of thin on the ground for a while. Read more!
Looks like a great line-up.
BTW, I'm really busy with outside projects right now, so big posts are going to be sort of thin on the ground for a while. Read more!
Friday, September 18, 2009
Energy and the Brain
The questions of how much energy is used by the brain, especially its various parts, and how it's used are important. For one thing, our understanding of the brain depends strongly on functional magnetic resonance imaging (fMRI), which in turn has a number of built-in assumptions and open questions regarding how blood flow and nutrient concentrations relate to energy usage within the tiny regions (voxels) that it can resolve.[7] [8] When dividing the brain into "parts" I'm talking not so much about areas or regions of the brain, as the microarchitectural constituents, such as axons, large and small dendrite branches, parts of the synapses on both sides of the synaptic cleft, and even astrocytes and other glial cells. (There's considerable debate regarding how much and what types of energy transfers take place between glial cells and neurons.[8])
Thus, a very recent paper in Science,[1] Energy-Efficient Action Potentials in Hippocampal Mossy Fibers (by Henrik Alle, Arnd Roth, and Jörg R. P. Geiger) provides an important resolution to an open question regarding energy usage in unmyelinated axons. They studied the current flows in axons of the Hippocampal Mossy Fibers, and demonstrated that the axons of these cells likely use about a third of the energy predicted by the standard notion, which is based on work going back to 1952.[13] [14] The general applicability of this notion has been disputed, however, since at least 1975 based on early data[11] on unmyelinated axons of different species obtained with radiolabeled K+.[1]
I'm going to start with the implications of this finding, followed by a discussion of what Alle et al. did and didn't discover, followed by a brief summary of what they did to perform this measurement.
Implications of the Lower Axonal Energy Usage
I've previously discussed the various functional aspects of the brain, in terms of performing the calculations (computations) leading to its function. These include the general system of action potentials (APs) being fired in neurons, traveling along the axons to the pre-synaptic areas where they stimulate the release of neurotransmitters, which cross the synaptic cleft to stimulate currents in the post-synaptic areas in dendrites of other neurons, which currents in turn produce voltage changes that are transmitted to the soma (neural cell body), the axon hillock, and the Axon Initial Segment (AIS) which are the most common locations for the firing of new APs (primarily the AIS). I've also discussed the ways in which many calculations can take place beyond the simple determination whether/when to fire an AP, as well as the non-linear ways in which the dendrite behaves as an "active cable", rather than the passive cable used in simpler models of neural activity.
Now, in order to behave as an "active cable", the dendritic membrane has to have some level of on-going current that can be modified in a non-linear fashion in response to voltage changes. These currents, or rather the ion-pumping activity required to maintain (or recover) the concentration gradients that drive them, cost energy just as do the currents in the synapses and axons. We have general ideas how much total energy any region of the brain uses during various activities, and the reduction of how much we think the small, local, unmyelinated axons are using means there's more left over for the other functions, including membranes with "active cable" characteristics. ...
The Results of the Research
Let's start with the easy stuff. This study was done in the hippocampus, which is one part of the brain out of more than a hundred. We can't know for sure that similar energy-efficiency holds in any other regions of the brain until similar studies have been made for them. Similarly, this study was done in rats, and in principle we don't even know if the findings hold true for mice, much less monkeys or humans. Finally, these findings apply to only one kind of cell in the hippocampus. In principal it might not hold for the other types of cell even there.
Realistically, however, it's reasonable to assume that what holds in one place holds in all, at least potentially. Various studies of brain energy have suggested much lower values for axon energy usage,[11] and we can assume that what evolution has done in one place, it can do in others, assuming some sort of selective incentive to reduce energy expenditure. And I think we can. (Ideally, there should be some scattershot studies of other cell types and regions, to verify the general principle. Hopefully this will offer opportunities for various researchers to get published, now that the cream has been skimmed off the discovery.)
Given the energy incentives for large-brained creatures, it seem likely that this energy efficiency evolved early in the lineages leading to mammals (and likely dinosaurs and birds as well, maybe independently). However, the rapid early expansion of the brain in Hadrocodium wui, to a point large even for modern mammals,[15] may represent the first opportunistic use of some mutation allowing for this energy efficiency. (Studies of monotreme, bird, crocodilian, and other reptilian (and perhaps amphibian, depending on reptilian results) axon current flows are strongly indicated.)
In general, then, unmyelinated axons in mammalian brains can probably be assumed to be as energy-efficient as their needs for high speed will allow. Further research and modeling will probably give us a good idea what the trade-offs are, this can be expected to be a hot area of research for a while.
Now, let's take a look at what, specifically, was discovered.
I've included links to several discussions of how action potentials work, so I'm not going to try to cover everything here. Basically, there are several ion flows involved in the action potential in the axon, but primarily they are sodium (Na+) and potassium (K+), with the Na+ concentration much higher outside the cell than inside, thus creating a current when it flows into the cell (INa), and the opposite for K+ (which currents are abbreviated Ik). These two currents are in opposite directions, and if they occur simultaneously at any one spot along the axon they will cancel out, while taking up energy.
In the earliest research into such currents, which were done in the giant axon of the squid,[13] [14] there appears to be considerable overlap. (This type of axon was used because its large size allowed researchers "to insert voltage clamp electrodes inside the lumen of the axon", even at this comparatively primitive stage of the technology.) The assumption was made that this overlap was general, even in mammals, although (as mentioned above) other research on unmyelinated axons suggested otherwise.[11]
As it turns out, Alle et al. have discovered that there's much less overlap of currents than previously assumed because the IK came mostly after the INa was complete. They also determined, through simulations, that
the observed degrees of charge separation are accompanied by comparatively low peak conductance densities, suggesting low numbers of channel proteins per area, which would minimize infrastructural costs for AP conduction.Thus, not only are APs cheaper in energy costs than has been assumed, but the cost of producing the infrastructure is also lower.
How the Research Was Done
Alle et al. used a technique called patch-clamp recording to measure the currents found in the membrane of rat hippocampal mossy fiber boutons (MFBs). In patch-clamp recording, a small section of cell membrane is removed with a pipette, in this case from boutons, which are small enlargements of the axon containing the pre-synaptic portion of synapses. A voltage command was applied that duplicated "a previously-recorded AP wave", and the currents were measured.
The onset of K+ currents (IK; Fig. 1, B and C, blue traces; n = 8) was significantly delayed compared to that of INa (106 ± 5 µs; P < 0.001), similar to results obtained from whole-bouton recordings (Fig. 1D, 115 ± 7 µs; P < 0.001, n = 8; P > 0.5 for patch versus whole-bouton recording). The resulting small overlap of inward and outward currents [Fig. 1, B (inset) and C] indicated a high Na+ efficiency and, accordingly, energy efficiency in hippocampal mossy fibers, contrasting with previous simulations of axonal APs and their underlying currents ([refs]).[1]Untangling the technical language, we see that the cell membrane of these particular axons responds to the voltage regime found in the AP with currents that barely overlap. This is the core finding.
There were also simulations:
To complement these results by a quantitative assessment of the Na+ influx as well as peak Na+ and K+ conductance densities (GNa and GK) underlying an AP propagating along an axon, we performed numerical simulations of APs. We used conductance functions (Fig. 2A) derived from recorded currents (Fig. 1) in a compartmental model of the mossy fiber ([ref]) to reconstitute propagating APs ([ref to supporting data]). Simulations resulted in AP waveforms and underlying currents closely resembling recorded APs and currents (Fig. 2B and fig. S1, A to D). The validity of our approach was further tested with independent predictions of the model, such as INa onset potential and AP propagation velocity, which both complied with experimental data (Fig. 2C and fig. S2).[1]These demonstrate that the values and timings of the currents involved, when incorporated into simulations, match the observed data.
They also analyzed the energy costs of the activity at the synapse that results from arrival of an AP, estimating that
the cost ratio of the mossy fiber AP itself to the downstream events (Fig. 4) has an upper limit of about 0.15 ([ref to supporting data]), shifting the emphasis of activity-dependent energy demand to downstream processes elicited by transmitter release, as suggested by in vivo work ([refs]).IOW the APs require less energy, so there's more for other processes.
Alle, H., Roth, A., & Geiger, J. (2009). Energy-Efficient Action Potentials in Hippocampal Mossy Fibers Science, 325 (5946), 1405-1408 DOI: 10.1126/science.1174331
Links: I've included only the links called out in this leader. Not all of these links are called out in the text. Many are references taken from the featured paper. Use the back key if you came via clicking a footnote.
1. Energy-Efficient Action Potentials in Hippocampal Mossy Fibers paywall
2. An Energy Budget for Signaling in the Grey Matter of the Brain Open Access
3. The neural basis of functional brain imaging signals
4. The Cost of Cortical Computation Open Access
5. Hemodynamic Signals Correlate Tightly with Synchronized Gamma Oscillations Free Registration Required
6. Coupling Between Neuronal Firing, Field Potentials, and fMRI in Human Auditory Cortex Free Registration Required
7. What we can do and what we cannot do with fMRI
8. Metabolic and hemodynamic events after changes in neuronal activity: current hypotheses, theoretical predictions and in vivo NMR experimental findings Open Access Author manuscript
9. An Energy Budget for the Olfactory Glomerulus Open Access
10. Functional Trade-Offs in White Matter Axonal Scaling Open Access
11. Energetic aspects of nerve conduction: The relationships between heat production, electrical activity and metabolism paywall
12. Cortical Action Potential Backpropagation Explains Spike Threshold Variability and Rapid-Onset Kinetics Open Access
13. The Optimum Density of Sodium Channels in an Unmyelinated Nerve paywall
14. A QUANTITATIVE DESCRIPTION OF MEMBRANE CURRENT AND ITS APPLICATION TO CONDUCTION AND EXCITATION IN NERVE may be open access, slow loading
15. A New Mammaliaform from the Early Jurassic and Evolution of Mammalian Characteristics Free registration required
Read more!
Tuesday, September 15, 2009
Scientia Pro Publica #11 is Up
here.
Nothing of mine in it, evidently my post Semantic Strait-Jackets in Science wasn't acceptable. As for why, I won't even guess, but it wasn't that late a submission.
But the line-up looks pretty good, I'd recommend a visit. Read more!
Nothing of mine in it, evidently my post Semantic Strait-Jackets in Science wasn't acceptable. As for why, I won't even guess, but it wasn't that late a submission.
But the line-up looks pretty good, I'd recommend a visit. Read more!
Encephalon #75 is Up...
Here.
Nothing from here on it (nothing good ready), but there still look to be some pretty good posts. Read more!
Nothing from here on it (nothing good ready), but there still look to be some pretty good posts. Read more!
Monday, September 14, 2009
Homeotic Mutationism
The guest post a while back by Dr. Filler brought up the issue of "adaptationism" vs. "homeotic mutationism". It seems to me that this issue is an example of the simplistic use of formulas in science (which I recently decried) where a more thoughtful approach would end up without the controversy.
Summarizing from most of the various papers I read (see Links), the foundation of "adaptationism" is the assumption that the genetic variation on which Darwinian selection operates involves such small increments of change that they appear continuous. Mutation as a source of variation operates (in this model) almost independently of selection, adding new variation to the gene pool, while natural selection operates against the entire existing pool variation by altering the relative distribution of different alleles for genes with variation. There also seems to me to be a tacit assumption that variation exists in any direction (within "morphospace", see here for a discussion of morphospace), so that anytime there is a selective incentive for evolution in a particular direction, it will happen.
(Afarensis pointed me to the 1996 book Adaptation edited by Michael Robertson Rose and George V. Lauder, as discussing the current "adaptationist" program. Evidently, the "adaptationism" discussed by Dr. Filler, and the papers mentioned above, is what Rose and Lauder define as the "Old Adaptationism". This is, by all appearances, dead. The "new adaptationism" takes some account of the ability of mutations to be "large", as well as accepting the the reality of developmental limitations on variation.)
A Recently Generated Example of Homeotic Mutation
I'm going to (briefly) discuss a recent paper documenting an artificial homeotic mutation[15], in which an entire homeotic gene was rendered non-functional, and the impacts to various developmental systems was analyzed. This is certainly not the first such experiment, however its recent provenance, and the widespread impacts to diverse systems including "thoracic, lumbar, and sacral vertebrae and in the pelvis, along with alterations in the bones and ligaments of the hindlimbs" and "a reduction in lumbar motor neurons and a change in locomotor behavior",[15] make it an excellent example of the sort of mutations that underlie the concept of "homeotic mutationism".
Before we start, however, let's take a look at genes and how they work. Wiki defines a gene as "the basic unit of heredity in a living organism." This is fine as a functional definition, but the history of research into the subject has provided some excess baggage: some deadwood that needs to be cleared away. ...
I'm not going to even summarize the process by which DNA sequences are translated into proteins. I've discussed this in past posts, and Wiki has a good description here (including the larger articles they link to).

Figure 1: Summary level cartoon of the "central dogma" regarding transcription and translation from DNA sequences to proteins. (From Wiki)
Specifically, the "central dogma" has often been taken to mean that a gene consists of the portion of DNA that codes for proteins. This isn't actually true, according to Wiki:
The central dogma of molecular biology was first enunciated by Francis Crick in 1958[ref] and re-stated in a Nature paper published in 1970:[ref]As you can see, this "dogma" applies only to the translation of DNA sequences into amino acid sequences in proteins. It says nothing about whether the gene is, by definition, limited to the protein-coding region.The central dogma of molecular biology deals with the detailed residue-by-residue transfer of sequential information. It states that information cannot be transferred back from protein to either protein or nucleic acid.
A Local and Temporary Definition of "Gene"
For purposes here, I'm going to use a limited definition of gene: only for protein-coding genes of Eukaryotes.A1 A gene is the DNA that codes for proteins, all the sequence that is transcribed with it, and all the control sequences that (in sum) determine when/whether that gene is going to be transcribed.
This is different from the more common (often unstated) definition of only the coding region. It includes all the introns that are transcribed to RNA and then removed by editing prior to the beginning of translation. It also includes all the control sequences by which its transcription is controlled. From the point of studying mutations, adaptation, and selection, this allows most of the mutations that affect development to be included within the gene, by definition.
Technically, a gene also should include the DNA sequences that, while not affecting transcription, are close enough to do so if they should mutate from a neutral sequence to one that impacts transcription.
Now, many genes code for proteins that perform simple metabolic and/or "housekeeping" functions within the cell. Others code for structural proteins, that are used to build the cellular skeleton. But the most important genes, from the point of view of controlling development, are those whose proteins contribute to the activation of other genes. The network of information handling these proteins are involved in constitutes a powerful and complex analog computer within the cell.
How do these proteins and their DNA interact? Basically, any protein that can interact on a sequence-specific basis with DNA is a Transcription Factor (TF). (Technically, TFs are enzymes, as are all proteins that can catalyze a chemical process of any sort. In addition, we should probably demand that the interaction with DNA have some significant effect.) TFs can work to enhance the rate of transcription, reduce it, suppress it entirely, or do any of the foregoing to the effects of other TFs. They do this by spending part of their time connected to the sequence(s) they act on, and while they do other parts of the molecule interact with other enzymes, creating a complex analog logic. (See my early post How Smart is the Cell? Part II: The Gene Activation network as an Analog Computer for a more detailed discussion of this, and links to more technical and peer-reviewed discussions.)
All this depends on the interaction between the TF and the "Transcription Factor Binding Sites (TFBS or "binding site") that interact with" the TF. Now, as I mention:
A TF can interact with many binding sites, and its activity with each site will be independent of the others, except that when it's present in relatively small amounts, there will be competition among sites for TF activity.The previous post was concentrating on how the gene activation network makes up a complex analog computer, but here I'm going to concentrate on the way the various types of mutation interact with this computer.
The interaction between a TF and its binding site depends on the specific sequence of DNA in the binding site. However, experiments with TF binding have shown that there are many sequences that will bind any particular TF, usually all very similar. By comparing these sequences, it's usually possible to find a consensus sequence that is very similar to all of them.
The consensus sequence will generally have the highest binding energy, that is it will stick tightest to the TF. However, other similar sequences may be able to bind to the TF, although with different behavior. A few example consensus sequences are found in table 2 from The Evolution of Transcriptional Regulation in Eukaryotes.[10] This means that when there are multiple binding sites with slightly different sequences, they will have different binding energies, and the activity will be different for the same concentration of TF. Note also that there can be multiple TF's with similar (but non-identical) consensus sequences, so that different binding sites may bind to different TF's depending on the relative concentrations of the TF's.
The effect of each TF concentration on transcription rate will be generally analog. Although a high enough concentration will saturate any particular binding site, producing full-bore transcription (assuming it's an enhancer), lower concentrations will cause each TF/binding site activity to perform an analog calculation.
The Effects of Point Mutations
Let's start with a simple point mutation, the replacement of one base in the DNA sequence by another. When this happens in a coding sequence, it may be "silent", coding for the same amino acid, or "neutral", coding for a different amino acid that, however, doesn't make much (or any) difference to the protein function, or it might have an important effect. In any event, it affects every interaction of the protein, one way or another (and to some extent or another). But a change to one of the binding sites only affects that specific interaction with the TF(s) involved. Many of these changes have a very small effect on binding energy (which, in turn, has a small effect on the transcription rate). Others have a larger effect, sometimes much larger, but only on the transcription rate of the one gene involved.
But what happens when there's a similar point mutation to the coding regions for a TF? Here, the mutation may have an effect on many, sometimes many hundreds of, interactions. If every binding site had the same sequence, the effect of a point mutation to the TF would be the same for all the "downstream" genes it affects. However, the actual sequences at the binding sites often vary, and a change to the TF could well increase the binding energy for some binding sites, while reducing it for others. Moreover, the magnitude of the change can also vary, with some being minor and others major. Finally, the results of these changes will (often) differ depending on how the specific transcription logic for each binding site fits into the overall computing network.
This, then, is the foundation for homeotic mutations. Not every mutation to a TF constitutes a homeotic mutation, but there are some TFs that are involved in hundreds (or even thousands) of transcription control operations, and when a mutation occurs to such a gene, the effects can be widespread, unconnected, and apparently random. This, specifically, is the type of mutation that Dr. Filler has hypothesized occurred when the ancestors of the Great Apes split off from other lineages around 20MYA.
Now, the number of bases involved in any TF/binding site interaction might be around 5-20. There are many possible mutations that could occur either to the binding site or the TF, but the effect of any change in binding energy will usually be limited to a single (scalar) change. The more different binding sites a TF interacts with, the more "dimensions" a mutation to the TF can make changes to.
Although the case is not a simple example, I should mention that several Hox proteins (the quintessential homeotic genes) act on the same consensus sequence, but with:
subtle, but distinct, preferences to DNA sites that contained variations of the nucleotides within the consensus motif. We further showed that Hox proteins varied in their relative affinities for DNA. These data demonstrate that closely related Hox proteins exhibit subtle differences in DNA binding specificities and affinities. These differences are likely to contribute to the selective interactions of Hox proteins with target DNA sites in vivo.[11]These aren't recent mutations, the hox genes have been evolving separately for over half a billion years, but they are highly conserved, with many specific genes (coding sequences) showing almost identical activity when mouse coding sequences are grafted into fruit flies in place of the native version, and vice versa.
What this shows, then, is that changes to homeotic genes can alter the binding affinity to a large number of different sequences, in different ways. A new homeotic mutation would probably be much "rougher" in its effects, at least until subsequent mutations to the binding sites "smoothed out" the effects, but such mutations could certainly occur even with a point mutation.
Effects of Other Types of Mutation on Homeotic Genes
Several other types of mutation need to be considered here. One type is where a section of code from another, related, gene gets grafted into the coding sequence in place of an original of similar length. This could well happen during DNA repair of a gene with many relatives, such as the Hox genes. (Specifically, the repair of a double-strand break through homologous recombination, which could potentially use a related gene as a template rather than the other copy of the original.)
Here, we potentially have the recombination of the part of the protein that interacts with one end of the consensus sequence with the part from another, related but not identical, protein that interacts with the other end. The result could be a gene with widely changed interactions with the various sequences it controls. Moreover, the "grafted in" section has undergone its own long evolution, creating its own system of binding energies with different sequence fragments. Although the mutation is "random" in the sense that a the new protein's affinities to the various sequences it controls will change in unpredictable ways, it's "random" within a very constrained space of possible changes.
Unlike point mutations, these mutations are capable of producing enormously organized suites of changes, although such suites will almost always be mal-adaptive. But not always, just almost always.
Another type of mutation also involves DNA repair, but this time instead of grafting in a homologous sequence from a related functional gene, the replacement sequence comes from a pseudogene originally created as a non-functional copy of this gene, which has been sitting in the genome accumulating random mutations for some time (ranging from very recent to somewhat older). The importance of this type of mutation is that several point mutations can accumulate without having any effect (because a pseudogene doesn't get expressed), until a short sequence is copied into the functional gene during DNA repair. As with most mutations, the very large majority would be mal-adaptive, but those that are viable can cross deep valleys in the "fitness landscape" that a working gene can't cross because any of the individual point mutations would be lethal or very mal-adaptive.
One more type of mutation must be considered, and that's a change to the control sequences of the homeotic gene itself. While such a change wouldn't affect the binding affinities between the homeotic gene and the downstream genes it affects, it could cause the gene to be expressed in places it wasn't previously, with all sorts of potential odd effects. Like the other types of mutation I've discussed, the vast majority of such mutations would probably be mal-adaptive, but the occasional adaptive one could produce homeotic effects.
There are other types of mutation that could produce potentially viable homeotic mutations, but the ones discussed are sufficient for an explanatory example.
An Artificial Example of a Homeotic Mutation
Now that we've examined just what a homeotic gene is, and how mutations to their protein coding can have such widespread effects, let's return to the example I mentioned above.[15] Axial and appendicular skeletal transformations, ligament alterations, and motor neuron loss in Hoxc10 mutants by Sirkka Liisa Hostikka, Jun Gong, and Ellen M. Carpenter.
The specific gene involved is named Hoxc10. We must note that in mammals the hox genes are actually defined in four separate families, the result of "the ancestral vertebrate genome being twice duplicated in its entirety", or at least one complete duplication and one duplication of both hox families.[16] Hoxc10 is part of the third ("c") family of hox genes, descended from the Hox10 gene of the original single string in the old pre-vertebrate chordates.
What Hostikka et al. did was create a version of this gene "producing a protein lacking the ability to bind to DNA."[15] In mice where the allele was homozygous (where both chromosomes had this inactive allele), results included the following:
The lack of a functional Hoxc10 homeobox causes several homeotic transformations in the axial skeleton (Table 1, Figures 3 and 4). Wild-type C57Bl/6 mice typically have 30 precaudal vertebrae, with the more caudal vertebrae organized in T13L6S4 pattern, with thirteen thoracic vertebrae, six lumbar vertebrae and four sacral vertebrae [refs]. Eighty percent of the wild-type mice examined in this study exhibited this pattern, with the remainder of the animals showing some mild variation in the shape of the L1, L6, or S1 segments; these types of variations are within the range of normal [ref]. Hoxc10 mutant mice also have 30 precaudal vertebrae, but the patterning of the vertebral column is altered. Most Hoxc10 mutant mice (33 of 34 examined) show a partial to complete transformation of the thirteenth thoracic vertebrae, precaudal vertebra 20 (PC20) into a lumbar identity, typified by the reduction or complete loss of the thirteenth rib. By definition, thoracic vertebrae are those vertebrae with attached ribs; hence loss of the thirteenth rib suggests a posterior transformation of the most caudal thoracic vertebra into a lumbar identity. The most caudal lumbar vertebra (PC25) often undergoes a similar transformation into a sacral (S1) identity (Figures 3 and 4). The sacrum in wild-type animals is typically comprised of four vertebrae. The first two vertebrae, S1 and S2, have butterfly-shaped, fused transverse processes. The next two sacral vertebrae, S3 and S4, have transverse processes that are not fused, with S3 having butterfly-shaped transverse processes and S4 having club-shaped transverse processes similar to those seen on more caudal vertebrae (Figures 2 and 3). In Hoxc10 mutant animals, the fourth sacral vertebra (S3* in Figures 3 and 4) exhibits an intermediate shape between a normal S3 and S4, and there are usually five sacral-like vertebrae, three fused and two free. This suggests an anterior transformation of the first caudal vertebra into a sacral identity. This transformation restores the register of the vertebral column and thus there is no overall loss of precaudal vertebrae. In combination, these alterations produce a T12(T13/L1)L5S5 pattern in 94% of mutant mice. Heterozygous mice show several intermediate patterns, most commonly T13L5S5 (31.5%) or T12(T13/L1)L5S5 (38.9%), suggesting dosage compensation. The transitional vertebra, defined as the most anterior vertebra to show a lumbar rather than a thoracic articulation between the pre- and postzygapophyses [refs] is normally the tenth thoracic vertebra (T10), whereas in the homozygous Hoxc10 mutants the transition occurs at the ninth thoracic vertebra (T9) (Figure 4).[15]In addition to the changes to vertebrae:
The bones in the hip, the ilium, ischium and pubis, are constructed from independent condensations that grow together after birth. All three bones meet at the acetabulum, but the ischium and pubis also meet at the ventral edge of the pelvis. These two bones are angled 45° from each other on the anterior, acetabular end, and are connected by a cartilaginous bridge where they meet on the posterior side. This bridge undergoes calcification during puberty, and the pelvic apparatus is normally fully fused by eight weeks of age. In Hoxc10 mutants, the cartilaginous bridge forms normally, but the calcification process is delayed, leaving a prominent seam or indentation on the posterior edge of the pelvis, dyssymphysis ischio-pubica. In all mutant adults the bones touch each other but even in the mildest cases, a seam is visible unlike in the controls (Figure 5A, B; Table 2). The C57Bl/6 background strain has been reported to have high incidence of dyssymphysis, especially in females [refs], however, somewhat newer reports have claimed only 30% incidence which was lost when C57Bl/6 mice were crossed to another strain [ref]. To minimize differences attributable to genetic background, wild-type sibling controls were used for all experiments.[15]The hindlimbs were also affected:
There are several alterations in hindlimbs and hip joints in Hoxc10 mutants. During embryonic and early postnatal development, there are no visible skeletal defects in the hindlimbs. However, by four to six weeks postnatally, a bony ridge develops along the anterior longitudinal line of the shaft of the femur (Figure 5). A likely cause for the formation of this femoral ridge is the presence of an ectopic branch of the iliofemoral ligament. The iliofemoral ligament is a sheet-like structure on the anterior side of the femoral neck, acting as a part of the synovial capsule. Normally, the ligament joins the ilium anterior to the acetabulum of the hip joint to the intertrochanteric line of the femur. During development, this ligament expresses Hoxc10 (Figure 2C-F). The ischiofemoral ligament connects the dorsal aspect of the ischium to the femoral neck. In the mutant, the ischiofemoral ligament is visibly weaker than in the wild-type mice, and attaches more dorsally to the medial side of the rim. The iliofemoral ligament, on the other hand, has two femoral connections in the mutants: one normal connection that attaches as a part of the synovial capsule into the femoral neck and another that attaches onto the anterior shaft of the femur (Figures 5 and 6). This second ectopic branch attaches to and is part of the anterior femoral ridge. The point of attachment varies from just distal to the intertrochanteric line to halfway down the femur, likely affecting the extent of ridge calcification that varies between animals and progresses with age. This phenotypic attachment site variation likely drives the differences in the shapes of the femoral ridges of the mutants from prominent to moderate, reflecting additional stresses on the bone. The femoral ridges are found in all the mutants six weeks and older (Table 2). Histological study of the extra ligament shows an organized fibrillar structure, indicating no aberrant pathology (Figure 6C). Serial sections of newborns show the ligament attaching to the femoral shaft (Supplemental Figure 2), also seen in some newborn skeletal preparations as Alcian blue-stained material in the ridge area indicating cartilaginous material, likely sulfated proteoglycans, at the femoral end of the abnormal ligament (Figure 5F, G).[15]There were also effects on the nervous system:
Initial analysis of peripheral nerve projection patterns in Hoxc10 mutant embryos using anti-neurofilament antibody labeling suggested that there were no gross defects in the formation, appearance, and projection of spinal nerves in Hoxc10 mutant embryos. Motor and sensory projections into the developing hindlimb bud appeared normal as well (Figure 2). However, in light of alterations in motor neuron positioning in paralogous Hox10 gene mutants [refs] we decided to examine these neurons more carefully in serial histological sections collected from newborn animals. Serial 10 µm sections were collected from three wild-type and three Hoxc10 mutant animals. Lumbar segmental position was established as previously described [ref] and the number of motor neurons in the medial and lateral motor columns (MMC and LMC, respectively) in the L1-L4 spinal segments was counted. All motor neuron populations showed substantial reductions in Hoxc10 mutants (Figure 8). We further distinguished both medial and lateral components of the LMC; both components showed similar reductions in numbers of motor neurons. Despite the reduction in numbers of motor neurons, the distribution of these neurons appeared relatively unchanged, with MMC and LMCM neurons showing a peak in their distribution in segment L2 and LMCL neurons peaking in segments L3/L4. This suggests an absence of an anterior spinal segmental transformation, in contrast to results observed in Hoxd10 mutants and Hoxa10/Hoxd10 double mutants [refs]. These cell counts appear to reflect an absolute loss of motor neurons, as there is no evidence of compensation in one pool for losses in another.[15]Note that these changes are the result of complete elimination of DNA-binding functionality. If the mutation had made small changes to the affinity for all the possible binding sites (rather than setting them all to zero) any or all of the areas found in this study might have been affected, but in different ways. Of course, as mentioned above, the hox genes (in mammals) are a complex family of multi-duplicated genes, with similarities in affinities between members of the same family. Thus, mutations to Hoxc10 will interact with Hoxa10 and Hoxd10 (there's no hoxb10), with the possibility for much more complex results.
The Adaptive Fitness of Homeotic Mutations
The subject gets sort of sticky here. As with most mutations, the ratio of "adaptive" to "mal-adaptive" mutations is usually very small. However, the number of possible mutations is also extremely tiny compared to the "morphospace" within which such developmental changes operate. This is especially true because any change to the protein coded by a homeotic gene will have a specific effect on each possible sequence used in a binding site. OTOH, in the case of a normal homeotic gene (as opposed to the complex families of Hox genes), the only thing that really matters for any specific binding site is the affinity its sequence has for the TF. There can potentially be a number of different sequences for the same binding site (multiple alleles of the same gene, using my definition of gene) with essentially identical affinities for the old TF, that would respond differently to the mutant version. Thus, closely related species could have several, or even many, binding sites with different sequences that all act identically (between species) with the old TF, but react differently to the same mutation.
Indeed, such an effect doesn't even require different species. Since a mutation to a specific binding site is "silent" if it doesn't affect the affinity for the old TF, even a small population could have several different versions of the same binding site, with different responses to the same mutation. Since the gene for the TF will often be on a different chromosome from the "downstream" genes the TF controls (including their binding sites, which are usually near the coding region on the same strand of DNA), the mutant form will undergo the full "shuffling" activity of sexual recombination.
Mutationism and Adaptationism
Now, lets take a look at how all this mixes with "adaptationism". Many point mutations to binding sites will have a very small effect, either because they have a naturally small effect on affinity, or because the logic of the control network causes even a large change to affinity to have a small effect on overall phenotype. This is probably the most common source of "variation" found in typical animal morphology. Mutations with larger effects will occur, but will have a much larger chance of being mal-adaptive and thus being selected out of the population.
An important aspect, however, is that the only variation that this type of mutation can produce in the phenotype is that which can be caused by changes to the affinity of one or more binding sites for their TF(s). Thus, the morphospace within which the typical variation resides is much "flatter", ie. it has many fewer dimensions, than the changes that can be imagined for observed morphology. This places limits on how evolution can adapt a population for changing conditions: if a particular position in morphospace can't be reached via changes to binding site affinities, the form involved won't appear in the species no matter how strong the selective incentive.
What mutations can occur, sooner or later do, assuming a large enough population. They are then tested for "fitness" within the population, with mal-adaptive mutations quickly disappearing (with the rare exception of one that happens to be linked to a highly adaptive one). However, as the probability of a mutation decreases, the size of a "large enough" population increases. When the critical size for a mutation to likely occur becomes enough orders of magnitude larger than the typical population integrated over the entire lifetime of the species,A2 the chance that that mutation will occur during the existence of the species in question becomes small, and the activity of evolution by natural selection becomes much less like the "adaptationist" model, and much more "mutationist", indeed, such mutations, many of which are homeotic, fit fairly cleanly into the old "hopeful monster" paradigm.
And it is here that we come to the point of the dispute. It can't be denied that many mutations produce very small changes in one or a few phenotypic characteristics. These, in the model I've discussed, will usually be changes to the control sequences rather than the amino acid sequence of the protein coded for. This means that the adaptationist models are certainly valid for evolution, both within species and to create new species. But the existence of homeotic mutations, and our modern understanding of how the various TFs and their binding sites combine with other aspects of cellular intelligence to drive the fetal development of the organism, make the "hopeful monster" paradigm just as valid, if in a substantially updated form.
When it comes to the many types of homeotic mutations that have such a low probability of occurring that there's, say, only a few percent chance of it happening during the lifetime of the species, we have pretty much left "adaptationism" behind, and are into the realm of "random creativity". In this type of scenario, the rare adaptive homeotic mutation can cause the origin of a new species, or higher level clade, if it occurs, but that occurrence is a matter of random chance. This is not to suggest that such mutations are in any way "directed", or that there are other forces than natural selection that determine the success of a particular homeotic mutation.A3 Rather, the "inevitability" of any particular homeotic mutation has been totally falsified (again, only in the case of the very low-probability ones), and the course of evolution is basically determined by the random chance of which such mutations arise when.
We see, then, that the "debate" between "adaptationism" and "mutationism" is either a matter of emphasis, or the denial of one or another valid mechanism by extremist proponents of the other. Both are valid, and technically even the most "hopeful monster" oriented form of homeotic mutationism still depends on natural selection and adaptation to pick out the rare adaptive mutation from the much larger mass of mal-adaptive.
Hostikka SL, Gong J, & Carpenter EM (2009). Axial and appendicular skeletal transformations, ligament alterations, and motor neuron loss in Hoxc10 mutants. International journal of biological sciences, 5 (5), 397-410 PMID: 19623272
Appendices:
A1. only for protein-coding genes of Eukaryotes: The differences between Eukaryotes and Prokaryotes include major differences in how they use DNA: in Eukaryotes the coding section of each gene is usually isolated along with its control sequences, so that different genes can reside on different chromosomes. In prokaryotes, most coding sequences tend to be combined into operons, with many genes being controlled by the same (set of) control sequences.
In addition, there are, in both types of life, genes that code for ribozymes (strands of modified RNA that function as catalysts in the manner of enzymes). These are often combined into operons even in Eukaryotes, so they don't fit into the logic of my argument here.
A2. the entire lifetime of the species: This makes sense at a high level, however when you dig into the details it gets a little iffy. For the typical species that lasts a few million years, with a population of millions to billions, if the probability of occurrence of a mutation is less than 1/total number of members (over the entire lifetime of the species), there will be significant chance that it will never occur. If it is much smaller, then the chance of its occurrence at all becomes small as well.
Where things get complicated is for species that have very large populations, especially divided into many distinct sub-populations, and that last a long time, say an order of magnitude longer than the typical few million years. Here, "silent" changes to various binding site sequences will have enough time to accumulate and diverge among sub-populations and over time, so that the exact same mutation to a homeotic gene will have different effects at different times and in different populations. From an adaptive standpoint, these would count as different mutations, although the change to the DNA of the homeotic gene itself is identical.
A3. other forces than natural selection that determine the success of a particular homeotic mutation: As it happens, there are a number of mechanisms by which one copy of a chromosome, with all the alleles it carries, can be preferentially selected during oogenesis or spermatogenesis.[14] I'm not going to discuss these here, although they cannot be ignored. See reference 14 for a more complete discussion.
Links: I've only included the link called out in this leader. Not all of these links are called out in the text. Use the back key if you came by clicking a footnote.
1. Adaptation from Leaps in the Dark Open Access
2. Mutationism and the dual causation of evolutionary change paywall
3. Climbing Mount Probable: Mutation as a Cause of Nonrandomness in Evolution paywall
4. Mutation-Biased Adaptation in a Protein NK Model Open Access
5. The concept of developmental reprogramming and the quest for an inclusive theory of evolutionary mechanisms paywall
6. WHAT IS EVOLVABILITY?
7. A framework for evolutionary systems biology Open Access
8. Saltational evolution: hopeful monsters are here to stay Open Access
9. The new mutation theory of phenotypic evolution Open Access
10. The Evolution of Transcriptional Regulation in Eukaryotes Open Access
11. Hox proteins have different affinities for a consensus DNA site that correlate with the positions of their genes on the hox cluster Open Access
12. Adaptationism by Peter Godfrey-Smith and Jon F. Wilkins (final draft: for the Blackwell Companion to the Philosophy of Biology. This draft has probably been peer-reviewed, but I'm not certain.)
13. Three Kinds of Adaptationism by Peter Godfrey-Smith (In S. H. Orzack & E. Sober (eds.), Adaptationism and Optimality, Cambridge University Press, 2001, pp. 335-357. This is not the published version, and may be prior to peer-review.)
14. Genes in conflict: the biology of selfish genetic elements By Austin Burt and Robert Trivers
15. Axial and appendicular skeletal transformations, ligament alterations, and motor neuron loss in Hoxc10 mutants Open Access
16. Extensive genomic duplication during early chordate evolution Open Access
Read more!
Thursday, September 3, 2009
Carnival of Evolution #14 is Up.
Here.
Unfortunately, I lost track of schedules (got several other things going on), and didn't get anything submitted, although there are two good candidates. I'll submit them for next month, and see what happens.
Meanwhile, the ones that did get there are pretty good, check out especially the one about how the appendix isn't vestigial after all. Read more!
Unfortunately, I lost track of schedules (got several other things going on), and didn't get anything submitted, although there are two good candidates. I'll submit them for next month, and see what happens.
Meanwhile, the ones that did get there are pretty good, check out especially the one about how the appendix isn't vestigial after all. Read more!
Saturday, August 29, 2009
Scientia Pro Publica 10: A Late Announcement
Find it here, unless you already did. (It's been there since the 19th.)
I kept a close watch for the first few days, then started to slack off. It was originally supposed to be hosted by Greg Laden at Quiche Moraine, and that was where I looked. Even after the announcement (on the 18th) that it would be published "tomorrow", I looked there. And didn't find it, and didn't find it, etc. It just never occurred to me to look back at Living the Scientific Life (Scientist, Interrupted) for updates, where I would have found the post itself.
If anybody was waiting for me to announce Scientia Pro Publica, sorry. But I suspect anybody really interested would have been Googling it, and found it before I did. But if you aren't interested, you should be. Lots of good stuff there. Read more!
I kept a close watch for the first few days, then started to slack off. It was originally supposed to be hosted by Greg Laden at Quiche Moraine, and that was where I looked. Even after the announcement (on the 18th) that it would be published "tomorrow", I looked there. And didn't find it, and didn't find it, etc. It just never occurred to me to look back at Living the Scientific Life (Scientist, Interrupted) for updates, where I would have found the post itself.
If anybody was waiting for me to announce Scientia Pro Publica, sorry. But I suspect anybody really interested would have been Googling it, and found it before I did. But if you aren't interested, you should be. Lots of good stuff there. Read more!
Thursday, August 20, 2009
Semantic Strait-Jackets in Science
While working on an earlier post, I ran into an annoying issue: in synapses, the result of a surge of GABA or Glycine is called and IPSP, meaning Inhibitory Post Synaptic Potential. This remains true even when the effect is actually excitatory. This is annoying, but easy enough to live with (for me) as I tend to think in historic terms, especially with regard to words.
But it brought to mind an interesting point: science is full of terms and definitions that are obsolete, with research subsequent to naming and definition having rendered the specific names, and/or some of the assumptions that went into them, invalid.
The case in point makes a good illustration: the assumption that a particular synaptic action in response to an action potential is inhibitory is built into the name, and it takes an effort of will and memory to keep track of the various times that it might be (or is known to be) excitatory.
But wait! There's more...
Not only does this semantic confusion result in lost time and errors when somebody forgets to allow for all the naming exceptions, but it also tends to channel everybody's thoughts into an assumption that the result of an action potential at a synapse is either one or the other, and that's it.
The reality, especially with chloride currents, is much more complex. ... Even if a burst of GABA causes the membrane to depolarize, the equilibrium voltage (also called the "reversal potential") may be somewhere between the resting voltage and the action potential threshold.

Figure 1: Effect of Cl- current with Equilibrium voltage at ~-62mV as an example. (Original. You may link to, copy, and or modify this image.)
In figure 1, we see just this situation. Compare with those from Axons and Chloride Currents, where the illustrated equilibrium voltages are either below the resting voltage (figure 2), or above the action potential threshold (figure 3).

Figure 2: Effect of Cl- current with Equilibrium voltage at ~-73mV as typical for a pyramidal cell soma. (From Axons and Chloride Currents. You may link to, copy, and or modify this image.)
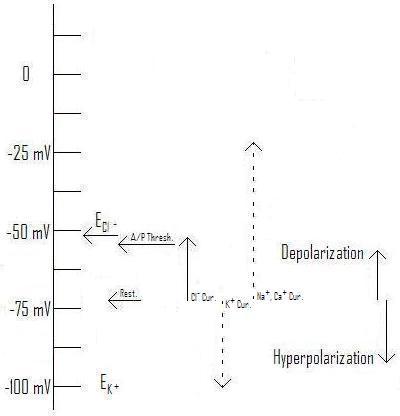
Figure 3: Effect of Cl- current with Equilibrium voltage at ~-54mV as typical for a pyramidal cell AIS. (From Axons and Chloride Currents. ; You may link to, copy, and or modify this image.)
In figure 1, a strong Cl- current will tend to depolarize the membrane, but only so far. If an EPSP based on sodium or calcium attempts to depolarize it further, the Cl- current will have a shunting action that will tend to resist this depolarization. How the membrane responds, and whether an action potential is fired, will be a very complex problem.
The very name IPSP, then, tends to prejudice our minds, hiding the complexity of what's going on. Ideally, IMO, this current should be renamed "CPSP" for "Chloride Post Synaptic Potential". This name is neutral with respect to whether it will tend to depolarize or hyperpolarize the membrane, as well as calling attention to the fact that the result is more complex than simply that binary choice.
Of course, I doubt it's going to change soon. Big Science has become as institutionalized as the Roman Catholic Church was in Galileo's day, if not as centralized in its power, and pushing such a change through would probably take as much effort as redefining Pluto as not a planet. Rather, this particular semantic strait-jacket will continue to inconvenience and distract people who haven't learned to live with it, along with all the other obsolete terms and definitions of this type.
The biggest problem I have with this isn't the distraction and tendency to hide important details. Rather, there are two ways to approach names, in science and anywhere else: you can remember them in functional terms, linking the words of the name to their everyday meanings, or you can remember them as arbitrary symbols, strings of words that represent a particular thing completely independently of their everyday meanings. When the string of words that makes up a scientific name doesn't represent the same thing as what the name is used for, only the latter way of remembering is available (unless you want to load up your brain with a bunch of the history of science). Thus, IPSP doesn't stand for an actual hyperpolarization of the membrane (which is what the words actually mean), but for a current carried by chloride or potassium ions, regardless of its effect on membrane voltage.
This turns learning about science into a process of memorizing pointless and arbitrary names, that can't be deciphered in terms of the everyday meanings of the words that make them up. Not only does this interfere with the education and development of people who would make good scientists, but it makes the field more attractive to the sort of people who don't do creative thinking: all they want to do is memorize formulas and rituals, and spend their career following the rituals they learned in college. Such people don't, IMO, really have much to contribute to science.
Read more!
But it brought to mind an interesting point: science is full of terms and definitions that are obsolete, with research subsequent to naming and definition having rendered the specific names, and/or some of the assumptions that went into them, invalid.
The case in point makes a good illustration: the assumption that a particular synaptic action in response to an action potential is inhibitory is built into the name, and it takes an effort of will and memory to keep track of the various times that it might be (or is known to be) excitatory.
But wait! There's more...
Not only does this semantic confusion result in lost time and errors when somebody forgets to allow for all the naming exceptions, but it also tends to channel everybody's thoughts into an assumption that the result of an action potential at a synapse is either one or the other, and that's it.
The reality, especially with chloride currents, is much more complex. ... Even if a burst of GABA causes the membrane to depolarize, the equilibrium voltage (also called the "reversal potential") may be somewhere between the resting voltage and the action potential threshold.
Figure 1: Effect of Cl- current with Equilibrium voltage at ~-62mV as an example. (Original. You may link to, copy, and or modify this image.)
In figure 1, we see just this situation. Compare with those from Axons and Chloride Currents, where the illustrated equilibrium voltages are either below the resting voltage (figure 2), or above the action potential threshold (figure 3).
Figure 2: Effect of Cl- current with Equilibrium voltage at ~-73mV as typical for a pyramidal cell soma. (From Axons and Chloride Currents. You may link to, copy, and or modify this image.)
Figure 3: Effect of Cl- current with Equilibrium voltage at ~-54mV as typical for a pyramidal cell AIS. (From Axons and Chloride Currents. ; You may link to, copy, and or modify this image.)
In figure 1, a strong Cl- current will tend to depolarize the membrane, but only so far. If an EPSP based on sodium or calcium attempts to depolarize it further, the Cl- current will have a shunting action that will tend to resist this depolarization. How the membrane responds, and whether an action potential is fired, will be a very complex problem.
The very name IPSP, then, tends to prejudice our minds, hiding the complexity of what's going on. Ideally, IMO, this current should be renamed "CPSP" for "Chloride Post Synaptic Potential". This name is neutral with respect to whether it will tend to depolarize or hyperpolarize the membrane, as well as calling attention to the fact that the result is more complex than simply that binary choice.
Of course, I doubt it's going to change soon. Big Science has become as institutionalized as the Roman Catholic Church was in Galileo's day, if not as centralized in its power, and pushing such a change through would probably take as much effort as redefining Pluto as not a planet. Rather, this particular semantic strait-jacket will continue to inconvenience and distract people who haven't learned to live with it, along with all the other obsolete terms and definitions of this type.
The biggest problem I have with this isn't the distraction and tendency to hide important details. Rather, there are two ways to approach names, in science and anywhere else: you can remember them in functional terms, linking the words of the name to their everyday meanings, or you can remember them as arbitrary symbols, strings of words that represent a particular thing completely independently of their everyday meanings. When the string of words that makes up a scientific name doesn't represent the same thing as what the name is used for, only the latter way of remembering is available (unless you want to load up your brain with a bunch of the history of science). Thus, IPSP doesn't stand for an actual hyperpolarization of the membrane (which is what the words actually mean), but for a current carried by chloride or potassium ions, regardless of its effect on membrane voltage.
This turns learning about science into a process of memorizing pointless and arbitrary names, that can't be deciphered in terms of the everyday meanings of the words that make them up. Not only does this interfere with the education and development of people who would make good scientists, but it makes the field more attractive to the sort of people who don't do creative thinking: all they want to do is memorize formulas and rituals, and spend their career following the rituals they learned in college. Such people don't, IMO, really have much to contribute to science.
Read more!
Axons and Chloride Currents
It's commonly assumed that synapses using GABA and glycine as neurotransmitters are inhibitory, that is when an action potential causes the release of (one or both of) these neurotransmitters (from the pre-synaptic side of the synapse) the result is a reduced chance that the post-synaptic cell will fire an action potential. However, this is not always true, there are several common circumstances under which these neurotransmitters appear to increase the chance of an action potential firing in the post-synaptic cell, as discussed in two recent papers to be discussed here. First, let's look at the mechanisms responsible for this reversal of the standard expectations, then I'll go over the two papers, then integrate the subject with some of my previous discussions regarding the transmission of analog data (besides action potential timing) along the first few hundred microns of the axon.
The role of Chloride Currents in Neural Communication
In a recent post I described the general relationship of various currents, ion channels, and voltage events (e.g. spikes and action potentials) in the neural cell membrane, but deferred discussion of chloride (Cl-) currents because of their complexity.
The key item regarding any ion-carried current is the equilibrium voltage (also often called the reversal potential): that voltage (across the cell membrane) at which the electrical pressure driving a specific ion in one direction exactly balances the concentration-driven pressure in the other. The equilibrium voltage is primarily determined by the relative concentrations on either side of the cell membrane, so a change to either concentration can produce a change to the equilibrium voltage, and different concentrations across different parts of the cell (membrane) will result in different equilibrium voltages. (They will also result in a net ion diffusion, along with an energy cost to maintain it: see below.)

Figure 1: Action Potential Threshold and Directions of Depolarization and Hyperpolarization Relative to Figure 3 of A New Integrative Theory for Cortical Pyramidal Neurons. (From Figure 4 of the same post.)
As you can see from figure 1, the typical equilibrium voltage for Cl- falls right into the range of typical "resting" voltages across the membrane. All of these are within about 40 mV of a typical action potential threshold, which is primarily determined by the nature of the specific voltage-gated Sodium (Na+) channels present in that piece of membrane. (See my discussion here for more detail.) Now, let's compare the effects of changing the Cl- concentration inside the cell from that of the soma (body) of a typical pyramidal cell to that of the axon initial segment[A1] (AIS: the first part of the axon after it leaves the axon hillock).
Figure 2: Effect of Cl- current with Equilibrium voltage at ~-73mV as typical for a pyramidal cell soma.[1] (Original. You may link to, copy, and or modify this image.)
Figure 3: Effect of Cl- current with Equilibrium voltage at ~-54mV as typical for a pyramidal cell AIS.[1] (Original. You may link to, copy, and or modify this image.)
As you can see, raising the equilibrium voltage for Cl- changes the direction of the current and its effect. Rather than being inhibitory (hyperpolarizing), the chloride current is excitatory (depolarizing), tending to make the post-synaptic cell more likely to fire an action potential. Notice in this illustration (Figure 3) the equilibrium voltage is higher than the threshold for firing an action potential, so enough chloride current can "do the job" independently of help from depolarizing influences coming from the dendrites. This has actually been observed in experiments.[1] [21]
As mentioned in A New Integrative Theory for Cortical Pyramidal Neurons, the normal concentration difference is just about enough to create an equilibrium voltage in the same range as the typical resting potential. This is because the concentration of Cl- outside the cell is greater than that inside by just enough to counteract the resting voltage. If, somehow, a higher concentration of Cl- is present on the inside of the membrane, the equilibrium voltage will rise, as shown in Figure 3 for the AIS.
Now, we can't just leave it that the inhibitory effects of neurotransmitters such as GABA and glycine can be reversed, we need to consider both the receptors (all types) and and the effects of changing the voltage under inhibitory conditions. ... (read the rest in the full post)
Let's start with Figure 2, when a big dose of GABA arrives at a synapse. The fastest-acting receptor(s) for GABA are the GABAA receptors, in which the molecule that spans the membrane contains both a pore through which the Cl- ions can pass, and a receptor region (outside the cell) that fits to GABA like a lock to a key, changing its shape (and the shape of the molecule), and allowing the pore to open. (A good discussion of this receptor may be found at The Versatile GABAa Chloride Channel Receptor Complex at Physiology physics woven fine. I'm not going to try duplicate this work, but in reading it just keep in mind that the author has not considered that the GABAA receptor can sometimes be excitatory.)
Notice, though, that the difference in voltages is very small. This isn't as important is it might seem, because when the depolarizing activity of an excitatory synapse (a regular one using i.e. glutamate or acetylcholine) tries to raise (depolarize) the voltage towards the threshold, the greater difference in voltage drives a greater current, tending to prevent the voltage from going very far from the resting potential.
By contrast, a potassium (K+) current will pull the voltage much farther down, distinctly hyperpolarizing the cell membrane, not just at the synapse but a considerable distance away. This will become important later (below), because another GABA receptor, the GABAB receptor, can cause K+ channels to open, having just this effect. Individual cells can express different mixes of receptors, and even the same cell can have different mixes in different parts of the cell.[27] [28]
This isn't just true of receptors, either. Ion pumps can be localized, with massively different densities in different parts of the cell membrane (as can many other membrane proteins.[27]
Now, let's look at the AIS, in Figure 3. Here, a big dose of GABA opens the pores in the GABAA receptor in the same way as above, but it produces a distinct depolarizing current, pulling the membrane voltage, as always, towards its equilibrium voltage, which in this case actually happens to be higher than the action potential threshold. Thus, in this case the GABA can not only be excitatory, it can trigger an action potential. (Of course, if the post-synaptic cell also expresses GABAB receptors, the effect will be much more complex: see below.)
Chloride Concentrations and Their Maintenance
What are the typical CL- concentrations driving these different currents? I'm not going to go into the extra-cellular (concentration outside the cell) concentration, since they're the same for all parts of the cell. (Since different parts of different cells are bathed in the same local extra-cellular fluids.) Instead we'll look at the intra-cellular (concentration inside the cell) found by Khirug, Yamada et al., one of the papers we'll be discussing.[20]
By determining the reversal potentials mediated by sudden release of GABA("EGABA values of –59.4 ± 1.5 mV (n = 14), –65.8 ± 1.2 mV (n = 14), and –70.9 ± 1.5 mV (n = 10), respectively (Fig. 1)"[20]), they could calculate the concentrations:
Assuming an intracellular pH of 7.2, the intracellular levels of Cl– ([Cli]) calculated on the basis of the above EGABA values ([refs]) are 11, 7.9, and 6.0 mM [milliMoles, a measure of concentration] for the AIS, soma, and dendrite, respectively.[20]Of course these values are typical of only one type of cell (dentate gyrus cells (DGCs) of the hippocampus of mice and rats), but they give us some numbers to work with.
How are these concentrations maintained against the inevitable leaks and currents, which would normally push ECl- towards the resting voltage?[20] [33] There are two specific transporters involved (out of a large number used throughout the body): Both are members of a large family called cation–chloride co-transporters (CCCs), transmembrane proteins that transport a specific mix of ions in a compulsory group (all together or none). One is a potassium-chloride co-transporter (KCC): KCC2, while the other is a sodium-potassium-chloride co-transporter (NKCC): NKCC1. (The numbers are confusing, the "1" has nothing to do with the "2": the body also has KCC1 and NKCC2, although they aren't relevant to this discussion.)
KCCs, including KCC2, transport one K+ and one Cl- together, which is plenty to maintain the type of potential seen in Figure 2. The K+ has a strong pressure "pushing" it out of the cell, and it will drag the Cl- along with it. (Remember that since these ions have opposite charges, they represent currents going in opposite directions.)
NKCCs, including NKCC1, move one Na+, one K+ and two Cl- together, which means they will go in the other direction because the very high pressure "pulling" Na+ into the cell will trump the pressure of the other three ions.
During development, most neurons express NKCC1, making GABAA currents excitatory, which appears to be necessary for proper neural development.[2] [20] Mature neurons appear to mostly express KCC2 in the soma and dendrites, which makes Cl- currents hyperpolarizing. There has been some controversy over how the higher concentrations in the AIS were maintained, with the standard explanation being the absence of KCC2 in the AIS.
GABAergic Depolarization of the Axon Initial Segment in Cortical Principal Neurons Is Caused by the Na–K–2Cl Cotransporter NKCC1
This paper, (by Stanislav Khirug, Junko Yamada, Ramil Afzalov, Juha Voipio, Leonard Khiroug, and Kai Kaila,) investigates the conflict between the point (noted above) that the inevitable leaks and currents would normally push ECl- towards the resting voltage, and the fact that Cl- currents in the AIS have been observed to be distinctly depolarizing, which means that the concentration must be maintained at a level such that ECl- is distinctly higher than the resting voltage. A simple absence of KCC2, which would push ECl- lower, wouldn't cut it.
First, by subjecting different parts of the same cell to bursts of GABA (with any GABAB receptors blocked using "CGP55845A [(2S)-3-[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)-(phenylmethyl)phosphinic acid]"[20]) and measuring the resulting equilibrium voltages, they demonstrated the different values of EGABA for the AIS, soma, and dendrites. This may reasonably be assumed to be close to the value for ECl-, with only the GABAA receptors working.
Then, they did identical experiments similar to the one described above with both wild type mice (WT) and mutants unable to express NKCC1 anywhere(NKCC1–/–). The results:
The WT neurons showed an axo-somatic ΔEGABA that was similar (5 mV) to the one described above in the Thy1–mGFP neurons with AIS, somatic, and dendritic EGABA values of –61.3 ± 2.4 mV (n = 14), –66.0 ± 1.6 mV (n = 22), and –71.0 ± 2.0 mV (n = 16), respectively. Notably, in identical recordings from the NKCC1–/– neurons, there was no axo-somatic EGABA gradient; the values for EGABA at the AIS and soma were similar, –70.5 ± 1.5 mV (n = 17) and –70.3 ± 1.5 mV (n = 18), respectively. However, a somato-dendritic ΔEGABA similar to that in the WT neurons was still observed (dendritic EGABA of –74.6 ± 1.7 mV; n = 15). These data indicate a key role for NKCC1 in the generation and maintenance of the chloride gradient that results in depolarizing GABA responses in the AIS of mouse DGCs and suggest that NKCC1 has no significant influence on dendritic EGABA. Interestingly, the above observations are not compatible with the idea that the somato-dendritic EGABA gradient is set by NKCC1 and KCC2. The putative transport mechanism underlying this gradient is not in the focus of the present work, but preliminary observations point to a role of a bicarbonate-dependent somatic transporter that accumulates chloride in the DGCs (our unpublished data). [20]They have thus demonstrated, for two types of cells, the DGCs and "rat neocortical layer 2/3 pyramidal neurons", that the excitatory (depolarizing) action of GABA synapses onto the AIS is probably caused by the expression and localized targeting of NKCC1. They've also discovered another effect evidently not caused by NKCC1: the difference in Cl- concentrations between the soma and dendrites. It'll be interesting to see what further research turns up in this direction.
Complex Events Initiated by Individual Spikes in the Human Cerebral Cortex
This paper (by Gábor Molnár, Szabolcs Oláh, Gergely Komlósi, Miklós Füle, János Szabadics, Csaba Varga, Pál Barzó, Gábor Tamás) reports intriguing results from "complex events triggered by individual action potentials in the human neocortical network." Unlike previous research, they used slices of human brain with part of the small-scale network intact, and applied single strong spikes to a single cell and observed the result.
Before I go on with this, let me save a lot of readers the effort of digging into the paper over ethical concerns with this blockquote:
All procedures were performed according to the Declaration of Helsinki with the approval of the University of Szeged Ethical Committee. Human slices were derived from material that had to be removed to gain access for the surgical treatment of deep-brain tumors from the left and right frontal, temporal, and parietal regions with written informed consent of the patients (aged 18–73 y) prior to surgery over the last 4 y.[21](Anybody wanting more information can use part of the above blockquote in a text search in the article, which is open access.)
I'm not going to spend further on the methods, or the details of the results, instead jumping to a more "high-level" discussion, starting with another blockquote:
Our results show that a single spike of a pyramidal cell in the human cortical microcircuit is capable of activating complex sequences of postsynaptic potentials lasting an order of magnitude longer than detected previously [refs]. The initiation and internally precise temporal structure of these event series appears to follow a stereotyped mechanism of spike-to-spike transmission traveling through a subset of synaptically connected neurons that seems to be conserved across several brain regions of the human cerebral cortex. The flow of downstream activation that follows the first-order spike of the trigger pyramidal cell is directionally controlled at two consecutive synaptic steps. Second-order spikes are triggered exclusively in GABAergic interneurons and not in pyramidal cells due to interneuron-selective EPSPs of enormous amplitude. In turn, second-order spikes in axo-axonic cells give rise to third-order spikes detected only in pyramidal cells, resulting in trisynaptic EPSPs in the network because axo-axonic cells do not innervate other GABAergic cells [refs]. Synchronized to the spikes in axo-axonic cells, second-order spikes in basket cells and possibly in other types of interneuron [refs] elicit hyperpolarizing effects reported here as disynaptic IPSPs.What does this mean? The human brain, (or at least the parts of the neocortex investigated,) is a very complex network of cells with positive (excitatory) and negative (inhibitory) effects, in which a single spike applied to a resting network ends up producing a stereotyped mix of these effects. Basically, there is a "cascade" (a bad analogy, but...) of action potentials, starting with the artificially triggered
- "first-order spike", followed by one or more
- "Second-order spikes" in "GABAergic interneurons", including axo-axonic cells which have excitatory GABAergic synapses on the AISs of pyramidal cells, as well as "basket cells and possibly in other types of interneuron" which have inhibitory GABAergic synapses on the somas and dendrites of pyramidal cells, followed in turn by
- "third-order spikes detected only in pyramidal cells, resulting in trisynaptic EPSPs [excitatory postsynaptic potentials] in the network",
One of the key points, for our purposes, is that the "axo-axonic cells do not innervate other GABAergic cells", they terminate only on the AIS of pyramidal cells.[34] This means that the very excitatory GABAergic synapses we've been discussing play an essential role in this network process.
Beyond Action Potentials
So far, we've covered the currents resulting from action potentials in GABAergic synapses terminating on the AIS. But regular readers here will probably recall The Analog Axon, where I concluded:
It's not a totally new discovery, of course, that some nerve cells use more analog types of communication than just action potentials.[refs] Sensory nerves especially, often make use of analog calculations. Nevertheless, except for the sensory margins of cognition, the central nervous system is usually pictured as communicating through action potentials that carry only their relative timing as information. Over long distances, this is probably true.This is particularly important for the pyrmidal cells involved in the "third-order spikes" mentioned above, where the axo-axonic input has set up varying chloride currents--varying both along the AIS and over time. These currents set the membrane voltage, often to a point substantially above (more depolarizing) the nominal "resting" voltage, with equally varying effects on the size, length, and shape of the action potential, in addition to their contribution to the probability and timing of firing the action potential in the first place. While over longer distances these variations will go away, short collaterals to nearby neurons will cary them. Studies on the fixing the membrane voltage at the soma have shown effects propagating over 400 microns (μ),[15], and we can probably add the length of the AIS to similar effects generated by voltage changes there. Similar distances for propagating analog effects have been found in presynaptic hippocampal mossy fiber boutons,[14] the output synapses from mossy fiber cells where:However, a great many of the connections in the brain, including the neocortex, arguably the most important part of the relative expansion of the human brain, are within a few hundred microns, the distance discussed here.
Excitatory presynaptic potentials result from subthreshold dendritic synaptic inputs, which propagate several hundreds of micrometers along the axon and modulate action potential–evoked transmitter release at the mossy fiber–CA3 synapse.[14]Note that these effects in the dendrites have to cross the soma before they can extend "several hundreds of micrometers along the axon", presumably similar subthreshold synaptic inputs landing on the AIS would propagate farther.
How many synapses are we talking about to be affected by these inputs? From the research mentioned above fixing the membrane voltage of the soma:[15]
Layer 5 pyramidal neurons, like other cortical neurons, give rise to a local high density of axonal connections to other pyramidal and non-pyramidal cells[refs] (Supplementary Figs 1, 4 and 7). Indeed, examination of the main axon and local axon collaterals of biocytin-filled layer 5 pyramidal cells from our slices revealed, on average, 155 (±79; n=14 cells) putative synaptic boutons within 0.5-mm of the cell body, and 269 (±152) putative synaptic boutons within the first 1mm(Fig. 4c and Supplementary Fig. 7). These values are probably a significant underestimate of local synaptic connectivity, owing to the cutting of axons and limitations of axonal staining using the slice technique (see Supplementary Figs 1 and 4).[15]This means that the AIS and short collaterals of each pyramidal cell can potentially act as an independent integrator of axo-axonic inputs, feeding a real-time analog output from hundreds of inputs[31] to hundreds of outputs[15] through the size, duration, and shape of action potentials, as well as sub-threshold changes that could affect the size of the transmitter release burst at the synapse. Moreover, these effects are tunable: addition of GABAB receptors to a synapse could produce a longer-term (50-300 ms) K+ current counteracting the depolarizing effect when many bursts of GABA arrive within a short time.
Like many other recent discoveries, these add yet more complexity, and potential calculating power, to the overall activity of the brain. This is an effect that will be very difficult to model in the more traditional neural networks, hammering home yet again how we must not limit our ideas of the brain's ability by what current models and neural networks can do. There's more where that came from, and the next decade will almost certainly see increasingly complex and powerful models of what the brain can do.
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., & Kaila, K. (2008). GABAergic Depolarization of the Axon Initial Segment in Cortical Principal Neurons Is Caused by the Na-K-2Cl Cotransporter NKCC1 Journal of Neuroscience, 28 (18), 4635-4639 DOI: 10.1523/JNEUROSCI.0908-08.2008
Molnár, G., Oláh, S., Komlósi, G., Füle, M., Szabadics, J., Varga, C., Barzó, P., & Tamás, G. (2008). Complex Events Initiated by Individual Spikes in the Human Cerebral Cortex PLoS Biology, 6 (9) DOI: 10.1371/journal.pbio.0060222
Appendix: You can use the back key to return to where you were, or click on the quoted text to return to the line with the footnote.
A1. "axon initial segment" There's no link to Wiki because, as of this writing, I couldn't find anything on Wiki discussing it. So herewith...
As mentioned above, the AIS is the first part of the axon, "from the narrow beginning of the axon to the onset of the myelin sheath" (often more than 200 microns (μ))[35], or in unmyelinated axons "a variable distance, 4-5 μ." It has recognizable features:[31]
The initial segment of the axon is unlike any other known process of the nerve cell, and in certain respects it is unlike any other part of the axon itself. In the idealized nerve cell it arises from the summit of a conical projection on the surface of the perikaryon, the axon hillock (Fig. 1). The surface of the axon hillock is bounded by a plasma membrane with the usual trilaminate structure, but near the apex of the hillock (Fig. 2) a thin, dark layer of finely granular material appears just beneath the membrane. In low-power electron micrographs this granular material gives the membrane a dense appearance, so that on superficial examination the plasmalemma seems to be reduplicated or thickened. But the undercoating is not really a part of the limiting membrane; instead, it is a thin layer of powdery densities, about 100 Å [Ångströms] thick, separated from the surface membrane by a clear interval of about 30 Å (Figs. 3 and 5). As its margins are not distinct, these measurements cannot be precise. The undercoating extends from the narrow beginning of the axon to the onset of the myelin sheath, where it ceases as abruptly as it began. In unmyelinated axons the undercoating extends for a variable distance, 4-5 μ. Where the axon originates from a dendrite, the undercoating has the same sudden onset and is accompanied by other changes (see below) in the internal structure of the process that signal the beginning of an axon.[31]The above text, and the figures it references, are in Reference 31, which is open access, although it's rather old.
Both the axon hillock and the AIS are distinct compartments within the cell, which can be independently targeted by distinct mixes of specific receptors, ion pumps, and all the other types of machinery the cell uses.[27] [28] [29] Ultrastructurally:
In the axon hillock (Fig. 2) microtubules collect into bundles of three to five or more, which funnel into the initial segment and run parallel with one another throughout its length. Large initial segments have five or six such fascicles, while small ones may have only one. The number of microtubules in a bundle varies considerably but is probably characteristic of the type of neuron. In the Deiters cells of the lateral vestibular nucleus (Fig. 5) and in the motor neurons of the spinal cord, the number of microtubules included in a fascicle is usually three to five, and in the pyramidal cells of the cerebral cortex (Fig. 6) the number can reach 22. In the Purkinje cell of the frog, Kohno (13) found from 6 to 25 microtubules in a bundle, but in the rat the maximumThese "synaptic boutons attached to the surface of the perikaryon at the axon hillock" and "the initial segment of the cerebral pyramidal cell" are at the center of the primary thrust of this post.
number we have found in this type of neuron is only 10-12.In longitudinal sections the fasciculated microtubules in the initial segment often appear darker than the single microtubules in the rest of the nerve cell and its processes. This appearance is due only partly to overlapping of the microtubules in a bundle within the thickness of the section. In addition, each microtubule is surrounded by a cloud of fine fibrillar material that contributes to the general density of the fascicle. In transverse sections it can be seen that the microtubules are arrayed close together in a curving and sometimes branching line (Figs. 3, 4, and 6). Single or isolated microtubules are rarely encountered in the initial segment. Favorably oriented transverse sections show that the microtubules within the fascicles are bound together by thin, dark crossbars or arms (Figs. 3 and 6).
The bundling of the microtubules ceases abruptly at the beginning of the myelin sheath. Whether they continue down the axons as isolated microtubules or are replaced by new tubules beginning in this region could not be determined from the sections that we examined.
Although the axon hillock and the beginning of the axon fail to stain with basic dyes, clusters and rosettes of ribosomes do occur in the axon hillock and, in diminishing quantities, throughout the length of the initial segment. Apparently they are not numerous or concentrated enough to produce a basophilia that is recognizable in the light microscope. The ribosomes are usually, but not always, associated with a tubule or two of the endoplasmic reticulum. At the beginning of the myelin sheath they disappear while the endoplasmic reticulum continues in its agranular form throughout the axon.
Other cytoplasmic components of the axon, the neurofilaments, the mitochondria, multivesicular bodies, and various vesicles, all pass into the axon from the axon hillock without undergoing any distinctive change in their appearance or aggregation. Near the apex of the axon hillock the mitochondria, neurofilaments, microtubules, and endoplasmic reticulum all assume a remarkably parallel orientation as they funnel into the narrow initial segment.
It is common to find synaptic boutons attached to the surface of the perikaryon at the axon hillock, but they are unusual on the surface of most initial segments. For example, in sections through the initial segments of some 60 different Purkinje cells only one synapsing bouton was found. In contrast, nearly every section through the initial segment of the cerebral pyramidal cell shows an attached bouton (Fig. 7). No examples have yet been encountered of initial segments studded with boutons like the dendrites and perikaryon of certain cells.
In a few cases in which the apposition between an axon terminal and the initial segment was caught in a favorable plane of section, it was possible to see that the typical undercoating of finely granular material was interrupted at such sites and that the surface of the axon reverted to the appearance it normally has in the internodal segments. Only at the location of the "synaptic complex" or "active zone" was there a deviation from the normal, here resulting from the aggregation of fine filamentous material that formed the postsynaptic density. The typical undercoating of the initial segment resumed beyond the margin of the apposing terminal. If a neuroglial process was inserted between the terminal and the axon, breaking the apposition, the typical undercoating reappeared beneath it. In our material the number of cases in which we could clearly follow both the pre- and postsynaptic membranes was too small to allow us to generalize this description with assurance.[31]
The AIS is the most common site for an action potential to begin, followed by the axon hillock. As the general dendritic excitement (depolarization) increases, the site often moves back, first to the soma, then sometimes to the proximal dendrite(s).[35]
Links: (I've only included the links called out in this leader.) Not all of these are called out in the text. Use the back key if you came via clicking a footnote above.
1. Excitatory Effect of GABAergic Axo-Axonic Cells in Cortical Microcircuits requires free registration
2. Cation–chloride co-transporters in neuronal communication, development and trauma paywall
3. Chloride is preferentially accumulated in a subpopulation of dendrites and periglomerular cells of the main olfactory bulb in adult rats paywall
4. Cluster Analysis–Based Physiological Classification and Morphological Properties of Inhibitory Neurons in Layers 2–3 of Monkey Dorsolateral Prefrontal Cortex Open Access
5. Output of neurogliaform cells to various neuron types in the human and rat cerebral cortex Open Access
6. A Population of Prenatally Generated Cells in the Rat Paleocortex Maintains an Immature Neuronal Phenotype into Adulthood paywall Is this one relevant?
7. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system paywall
8. A subset of periglomerular neurons in the rat accessory olfactory bulb may be excited by GABA through a Na+-dependent mechanism paywall
9. Channel behavior in a gamma-aminobutyrate transporter Open Access
10. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance Open Access
11. Structure of intraglomerular dendritic tufts of mitral cells and their contacts with olfactory nerve terminals and calbindin-immunoreactive type 2 periglomerular neurons paywall
12. Compensatory changes in cellular excitability, not synaptic scaling, contribute to homeostatic recovery of embryonic network activity paywall
13. Enigmatic Central Canal Contacting Cells: Immature Neurons in "Standby Mode"? paywall
14. Combined Analog and Action Potential Coding in Hippocampal Mossy Fibers Requires free registration
15. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential
16. Long-Term Activity-Dependent Plasticity of Action Potential Propagation Delay and Amplitude in Cortical Networks Open Access
17. Information processing by graded-potential transmission through tonically active synapses
18. Enhancement of presynaptic neuronal excitability by correlated presynaptic and post-synaptic spiking
19. Recurrent excitation in neocortical circuits paywall
20. GABAergic Depolarization of the Axon Initial Segment in Cortical Principal Neurons Is Caused by the Na–K–2Cl Cotransporter NKCC1 Open Access
21. Complex Events Initiated by Individual Spikes in the Human Cerebral Cortex Open Access
22. Axonal GABAA receptors paywall
23. Robust Short-Latency Perisomatic Inhibition onto Neocortical Pyramidal Cells Detected by Laser-Scanning Photostimulation paywall
24. Excitatory GABAergic Activation of Cortical Dividing Glial Cells paywall
25. GABA Transporter GAT1 Prevents Spillover at Proximal and Distal GABA Synapses Onto Primate Prefrontal Cortex Neurons paywall
26. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations
27. Compartmentalizing the neuronal plasma membrane from axon initial segments to synapses paywall
28. Localization and Targeting of Voltage-Dependent Ion Channels in Mammalian Central Neurons paywall
29. The distribution and targeting of neuronal voltage-gated ion channels
30. Excitatory effects of GABA in established brain networks paywall
31. The axon hillock and the initial segment Open Access
32. Proximity of excitatory and inhibitory axon terminals adjacent to pyramidal cell bodies provides a putative basis for nonsynaptic interactions paywall
33. The cellular, molecular and ionic basis of GABAA receptor signalling
34. Salient features of synaptic organisation in the cerebral cortex
35. From molecules to networks : an introduction to cellular and molecular neuroscience edited by John H. Byrne, James Lewis Roberts, chapter 17
Read more!
Subscribe to:
Posts (Atom)